Sometimes all you need is to cut adapters
Published on
When you see a FastQC box like this, you know that something is wrong.

But what exactly?
Overrepresented sequences may indicate adapters (although, ironically, the Adapter Content is one of the few green check marks here), and duplicated sequences could be removed, but what do you do about the GC levels, per base sequence content, and per tile quality bias?
Here’s the table of overrepresented sequences identified by FastQC:
| Sequence | Count | Percentage | Possible Source |
|---|---|---|---|
| GATCGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTAT | 17953666 | 21.724791264966143 | TruSeq Adapter, Index 8 (97% over 36bp) |
| ATCGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTATG | 3032064 | 3.6689419031198587 | TruSeq Adapter, Index 8 (97% over 35bp) |
| GTTCGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTAT | 161627 | 0.19557637074136738 | TruSeq Adapter, Index 3 (97% over 34bp) |
| AATCGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTAT | 117432 | 0.14209831506431633 | TruSeq Adapter, Index 3 (97% over 34bp) |
| ATCGGAAGAGCACACGTCTGAACTCCCGTCACTCTCGCGCATCTCGTATG | 98036 | 0.11862823093914193 | TruSeq Adapter, Index 3 (96% over 33bp) |
| GATAGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTAT | 82961 | 0.10038676268862615 | TruSeq Adapter, Index 3 (97% over 34bp) |
Examining the reads further clarifies the picture: it’s not just that 25% of reads contain adapter sequences; most of them are adapter sequences right from base 1.
This can already explain a lot of what we’re seeing. If a quarter of the reads are variations of the same fixed string, it will definitely skew the GC content and the per base sequence content distributions. This also explains the tile bias — there was some issue with the library that was localized to a couple of lanes.
But the most miraculous thing is how trimming/removal of these adapter sequences affects the quality score distribution. Even if part of the reads were sequenced from adapter DNA instead of endogenous DNA, you wouldn’t necessarily expect those reads to be of lower quality, would you?
Here’s the per-base sequence quality in the raw data:
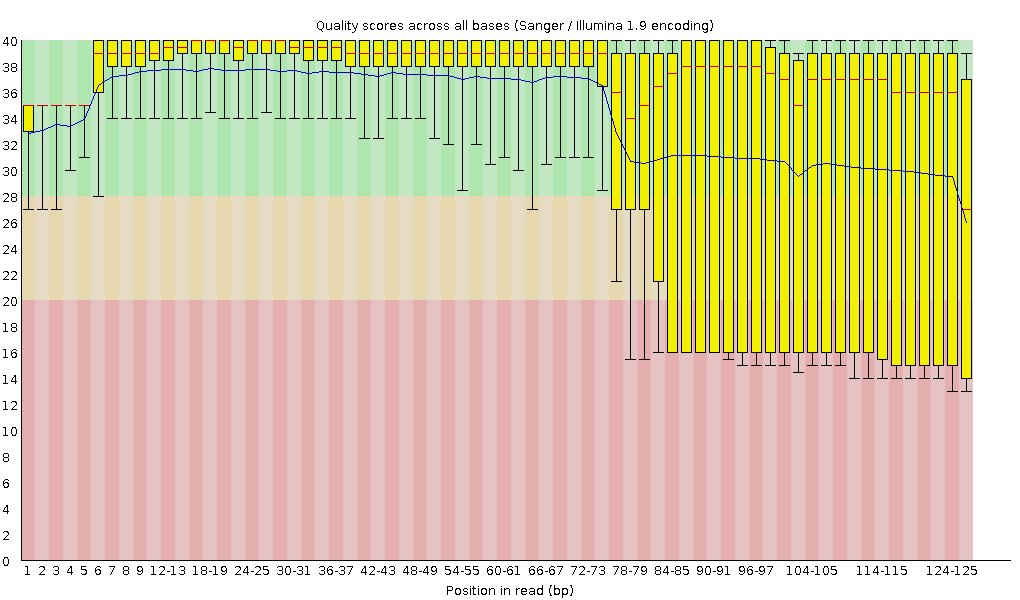
The analysis that I’m doing (ChIP-seq peak calling) is not very sensitive to the read length, so I was thinking about trimming the reads to 75bp or so just to avoid those questionable bases at the end.
But look what happens if I simply remove the adapters, without doing any other trimming or quality filtering:
I don’t quite understand why this happens, but a couple of explanations come to mind:
There’s a stretch of
As in the second half of the adapter sequence; e.g.GATCGGAAGAGCACACGTCTGAACTCCAGTCACTCTCGCGCATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAACACGAACATGCCGGTTGAAGCAAGTCTCCTGTATCTGGCCTCCACC ^^^^^^^^^^^^^^^So it could be this poly(A) segment that leads to errors.
There could also be a common issue that caused both adapter-only fragments and low quality scores, although I don’t have any ideas about what it could be.
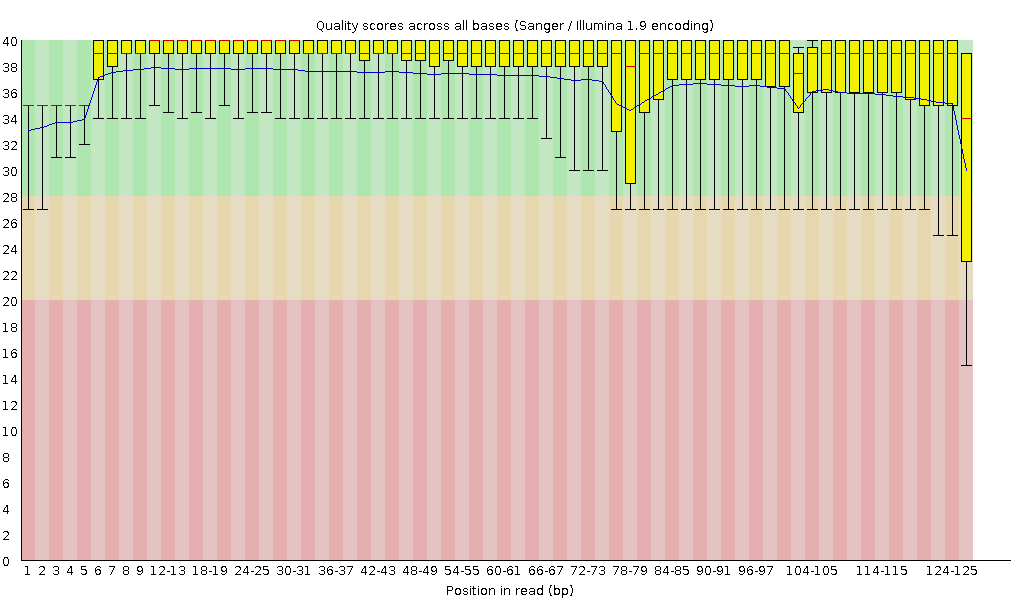
In any case, simply trimming the adapter sequence and removing all reads shorter than 40bp fixed pretty much everything:

The only remaining warning simply tells us that not all sequences are of the same length, as we would expect after trimming adapters:
This module will raise a warning if all sequences are not the same length.
[…]
For some sequencing platforms it is entirely normal to have different read lengths so warnings here can be ignored.
For the record, the command I used was
cutadapt --cores={threads} -a GATCGGAAGAGCACACGTCTGAACTCCAGTCAC -o {output} {input} --overlap 4 --minimum-length 40You can also browse and compare the full FastQC reports before and after adapter trimming.